Molecular crystal structures pack it in
 (Download Image)
(Download Image)
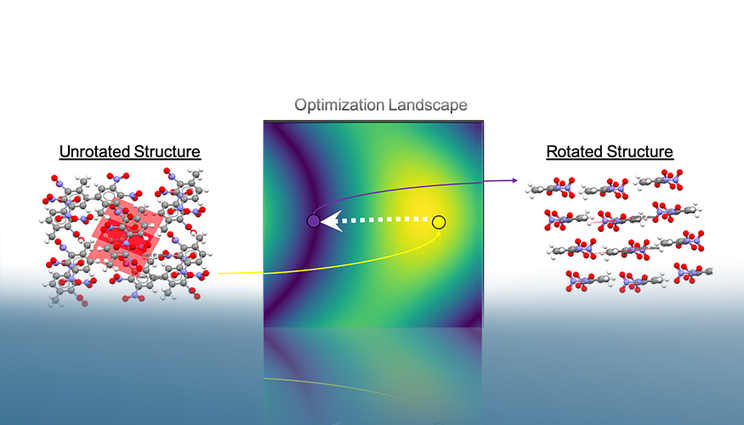
Autopack rotates crystal structures in 3D space to minimize their molecules’ projected area. After convergence, it is possible to extract the crystal’s associated packing motif based on relative interplanar angles. In this example, the stacks found after the optimization procedure indicate the structure’s beta packing motif.
Whether organic chemists are working on developing new molecular energetics or creating new blockbuster drugs in the pharmaceutical industry, each is searching how to optimize the chemical structure of a molecule to attain desired target properties.
Part of that optimization includes a molecular crystal’s packing motif, a perceived pattern in how molecules orient relative to one another within a crystal structure. The current packing motif datasets have remained small because of intensive manual labeling processes and insufficient labeling schemes.
To help solve this problem, a team of Lawrence Livermore National Laboratory (LLNL) materials and computer scientists have developed a freely available package, Autopack, which formalizes the packing motif labeling process and can automatically process and label the packing motifs of thousands of molecular crystal structures. The research appears in the Journal of Chemical Information and Modeling.
Small-scale crystal engineering studies over the past 30 years suggest that, while predicting experimental crystal structures from a chemical structure alone remains out of reach, there may be relationships between molecules’ chemical structures and a specific attribute of the crystal structure they adopt called the packing motif.
A molecular crystal’s packing motif is an important concept for energetics and organic electronics applications due to observed correlations between molecular crystals’ packing motifs and performance properties of interest, which include insensitivity for molecular explosives and charge transport for molecular semiconductors.
No formalized and open-source method of assigning packing motifs has ever been created until now. Instead, packing motifs are ascribed to molecular crystals simply by human evaluation of a crystal structure and judgment, resulting in small and noisy datasets.
“In the era of machine learning, the ability to create large, labeled datasets of molecular crystal packing motifs is now especially important,” said LLNL data scientist Donald Loveland, lead author of the paper. “Such efforts may generate models that can predict packing motifs from molecules’ chemical structure alone, which would help organic chemists prioritize syntheses of new molecules based on the desired packing motif and properties.”
The new LLNL work uses an efficient optimization algorithm that circumvents many problems found in previously proposed packing motif labeling methods, leading to new state-of-the-art results when tested on an LLNL-curated dataset.
Through Autopack, researchers have been able to generate a dataset of nearly 10,000 packing motifs for a set of energetic and energetic-like molecules of interest to the Lab, a task that would have been impossible before. For context, previous literature has remained capped on the order of 100 molecules due to the tedious and time-consuming nature of hand labeling. Early analysis of this new dataset hints at complex trends between intermolecular interactions, 3D molecular conformations and adopted packing motifs currently unexplored in the field, providing guidance on next steps for crystal engineering pipelines.
The code is freely available through the Lab’s Innovations and Partnerships Office.
Co-authors of the work include Bhavya Kailkhura, Piyush Karande, Anna Hiszpanski and Yong Han.
This work was funded by LLNL’s Laboratory Directed Research Development program.
Contact
 Anne M. Stark
Anne M. Stark
[email protected]
(925) 422-9799
Related Links
Journal of Chemical Information and ModelingAutopack Code
Tags
Advanced Materials and ManufacturingMaterials Science
HPC, Simulation, and Data Science
Computing
Physical and Life Sciences
Industry Collaborations
Technology Transfer
Featured Articles