LLNL’s new machine learning platform generates novel COVID-19 antibody sequences for experimental testing
 (Download Image)
(Download Image)
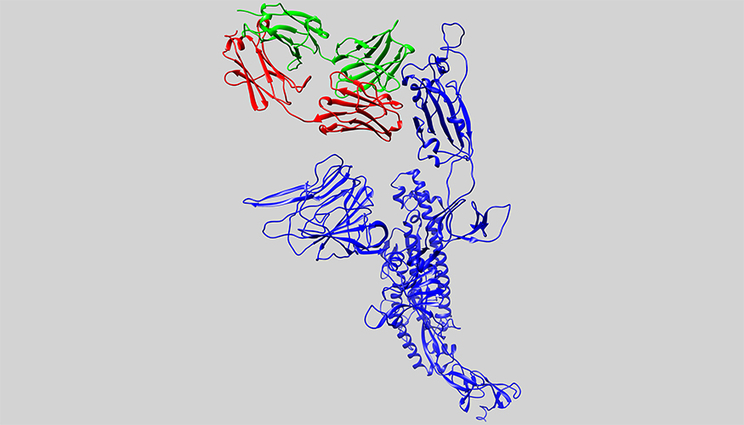
In this computer model, the 3D structure of an antibody candidate is shown alongside the protein of SARS-CoV-2, the virus that causes COVID-19. The key protein from SARS-CoV-2 (the spike protein) is shown in blue. The antibody (in green and red) binds and neutralizes SARS-CoV-1, but it doesn’t bind to SARS-CoV-2. It is the starting point for researchers to design an antibody to the virus that causes COVID-19.
Lawrence Livermore National Laboratory (LLNL) researchers have identified an initial set of therapeutic antibody sequences, designed in a few weeks using machine learning and supercomputing, aimed at binding and neutralizing SARS-CoV-2, the virus that causes COVID-19. The research team is performing experimental testing on the chosen antibody designs.
Currently, treating COVID-19 with antibodies is only possible by harvesting them from the blood of patients who have fully recovered. As the new antibody designs are improved through an iterative computational-experimental process, they could enable a safer, more reliable and scalable pathway to using antibodies as potential treatments for people stricken with the disease, scientists said.
In a paper appearing on the open access preprint website BioRxiv — which has not been peer-reviewed — LLNL scientists describe how they used the Lab’s high performance computers and a machine learning-driven computational platform to design antibody candidates predicted to bind with SARS-CoV-2 Receptor Binding Domain (RBD). Combining known antibody structures for SARS-CoV-1 (a similar coronavirus that causes Severe Acute Respiratory Syndrome) and a machine learning algorithm that proposed mutations to those structures to optimize them for SARS-CoV-2, Lab scientists whittled down the number of possible designs from a nearly infinite set of candidates to 20 initial sequences predicted to target SARS-CoV-2.
Free energy calculations performed for the first set of designs — used to predict the likelihood of binding — compared favorably to similar calculations for known SARS-CoV-1 antibodies, the team reported. The scores indicate that the predicted SARS-CoV-2 antibodies may bind to the virus’ receptors and neutralize it by preventing the virus from binding with and entering human cells. The antibody mutants also scored well on multiple developability metrics, meaning a high likelihood that they could be developed in a lab, researchers said. LLNL researchers have obtained the antibodies they selected and are performing real-world experiments in a lab setting.
“Our computational results are encouraging, and we’re excited about the experimental tests that are underway now,” said LLNL data scientist Dan Faissol, program lead for AI-driven vaccine and antibody design. “We hope that one of these initial antibody designs binds to the SARS-CoV-2 target as intended, but regardless of the outcome, the experimental results will significantly improve our ability to design a subsequent round of antibodies.”
Lab researchers said the sequences and binding calculations being made available to the scientific community also could help outside groups compare human-derived antibodies with LLNL’s free energy calculations to help choose which ones are worth pursuing further.
As previously reported, LLNL scientist and co-author Adam Zemla used the known protein structure of SARS-CoV-1 to produce a predicted 3D protein structure of SARS-CoV-2. Subsequently, the actual spike protein structure of SARS-CoV-2 was determined, demonstrating the prediction was accurate.
“In our effort to model the binding of SARS-CoV-2 with SARS-CoV-1 neutralizing antibodies, we knew that despite the high level of similarity between the two viruses, the SARS-CoV-1 antibodies do not bind to SARS-CoV-2,” said Zemla. “So, a real challenge in constructing our models is to predict specific structural changes present in SARS-CoV-2 that would thereby allow us to predict modifications to the antibody to compensate for those changes and establish binding.”
In just 22 days, using the SARS-CoV-2 protein sequence and known antibody structures for SARS-CoV-1, a Lab team led by Faissol and data scientist Thomas Desautels used a computational platform combining machine learning, bioinformatics, experimental data, structural biology and molecular simulations to drastically narrow down the possible antibody designs predicted to target SARS-CoV-2. The team used more than 200,000 CPU hours and 20,000 GPU hours on two high-performance computers at LLNL, Corona and Catalyst, to perform nearly 180,000 free energy calculations of candidate antibodies with the SARS-CoV-2 Receptor Binding Domain (RBD), according to the paper.
“The combination of all of these computational elements, including bioinformatics, simulation and machine learning, means that we can flexibly and scalably follow up on mutants with promising predictions as they emerge, effectively using Livermore’s HPC systems,” said Desautels, principal investigator on the project. “Searching a design space of this size just wouldn’t be feasible for an unaided human because the volume of decisions is too high.”
With their predicted SARS-CoV-2 structures, the team then used their design platform to computationally estimate the binding properties of almost 90,000 mutant antibodies. They selected the most promising initial antibody sequences and calculated them to have improved interaction with the SARS-CoV-2 RBD with free energies as low as -82 kilocalorie per mole, a unit of measure reflecting the strength of binding. The lower the number, the better chance of a binding match. For comparison, the figure for SARS-CoV-1 and one of its known antibodies is -52 kcal/mole.
“What we wanted was to get a figure that was at least as good as SARS-CoV-1, but for SARS-CoV-2, and we did, at least computationally, which was not an easy task.” Faissol said.
The LLNL team is performing additional calculations with different kinds of antibodies known to bind to SARS-CoV-1 and are continuing to improve the platform. They also are running higher fidelity molecular dynamics calculations to increase the accuracy of predictions and further investigating binding “hotspots,” where binding is dominated by a small number of contacts.
“Our initial platform leveraged free-energy calculations utilizing a common molecular modeling method that was a reasonable compromise between speed and accuracy,” said computational chemist and co-author Ed Lau. “We are now using a more accurate but computationally intensive method to calculate the binding free-energy of our new antibody designs.”
Lau said the team is performing these high-fidelity simulations based on a methodology developed from a collaboration between LLNL and Harvard University, which was supported by internal Laboratory Directed Research and Development (LDRD) funding.
The virtual design platform was supported by the Defense Advanced Research Projects Agency (DARPA) and LDRD funding. Researchers said aspects of the platform, including details on the machine learning model, the feature space representing the antigen-antibody interface and the antibody optimization pipeline, will be described in greater depth in a future publication.
Co-author Magdalena Franco, an LLNL biomedical researcher and a member of the antibody design team, is leading follow-on experimental efforts.
As part of its biosecurity mission, LLNL is committed to applying its leadership computing facilities as well as select experimental capabilities to help inform and accelerate the search for effective medical therapeutics.
For more on LLNL’s COVID-19 research, visit the web.
Contact
 Jeremy Thomas
Jeremy Thomas
[email protected]
(925) 422-5539
Related Links
LLNL’s COVID-19 research"Lawrence Livermore researchers release 3D protein structure predictions for the novel coronavirus"
Animation
Tags
Bioscience and BioengineeringBiosciences and Biotechnology
HPC, Simulation, and Data Science
Computing
Physical and Life Sciences
Emergency
Featured Articles