Understanding the spread and mutation of Zika virus
 (Download Image)
(Download Image)
Mosquitoes are responsible for the spread of the Zika virus. Credit: James Gathany/Wikimedia Commons
A team of Lawrence Livermore National Laboratory (LLNL) and University of California, Davis researchers has found that outbreaks of human disease, such as the 2015 Zika virus epidemic, may be due to genetic mutation, and viruses may undergo further changes as they expand their geographic range.
The question of how Zika virus (ZIKV) changed from a seemingly mild virus to a human pathogen capable of microcephaly (a type of neonatal malformation) and sexual transmission remains unanswered. The unexpected emergence of ZIKV’s pathogenicity and capacity for sexual transmission may be due to genetic changes, and future changes in phenotype may continue to occur as the virus expands its geographic range.
First isolated in 1947 from a rhesus macaque in Uganda, Zika virus was largely ignored until 2015 when a large outbreak of ZIKV infections in Brazil was linked to a dramatic increase in microcephaly cases. ZIKV has a number of unexpected attributes for an arthropod-borne virus (“arbovirus”). In addition to the unusual propensity of ZIKV to cause microcephaly and other birth defects, ZIKV can be transmitted sexually and has been shown to persist in the male and female reproductive systems.
To better understand viral evolution, the research team studied how the Zika virus mutated over space and time during the 2015 outbreak.
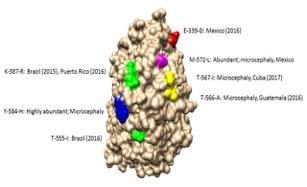
Model of the Zika virus NS3 protein that mutated during the 2015 epidemic with mutated regions highlighted. Several of the mutations occur in the exposed region of NS3 that interfaces with NS5 polymerase protein and may impact replication. Coloring scheme shows hydrophilic regions (blue) to hydrophobic regions (red).
“The sheer size of the 2015-16 epidemic may have brought attention to a pre-existing virulent ZIKV phenotype in a highly susceptible population,” said LLNL biologist Monica Borucki, lead author of a paper appearing in PLOS ONE. “It’s important to identify patterns of genetic change that may yield a better understanding of ZIKV emergence and evolution.”
ZIKV has an RNA genome and as is the case with all viruses with RNA genomes, ZIKV mutates rapidly due to its highly error prone RNA-dependent-RNA-polymerase. The high mutation rate of RNA viruses increases the ability of these viruses to adapt to diverse hosts and cause new human and zoonotic diseases. Specifically, ZIKV undergoes rapid mutation, which makes it difficult to identify combinations of mutations associated with viral emergence.
As next-generation sequencing technology has allowed whole genome consensus and variant sequence data to be generated for numerous virus samples, the task of analyzing these genomes for patterns of mutation has become more complex.
“Understanding which combinations of mutations spread widely and become established in new geographic regions versus those that disappear relatively quickly is essential for defining the trajectory of an ongoing epidemic,” said LLNL biologist Nicole Collette, a co-author of the study.
The researchers conducted a multiscale analysis of the wealth of publicly available genomic data generated over the course of the epidemic combined with in vivo laboratory data and were able to assess the trends in mutations and outbreak trajectory.
Mutations were detected throughout the genome via deep sequencing, and many variants appeared in multiple samples and in some cases became the consensus. Amino acids that were previously consensus in pre-outbreak samples were detected as low frequency variants in epidemic strains. Protein structural models indicate that most of the mutations associated with the epidemic transmission occur on the exposed surface of viral proteins.
At the macroscale level, consensus data was organized into large and interactive databases to allow the spread of individual mutations and combinations of mutations to be visualized and assessed for temporal and geographical patterns.
“The use of multiscale modeling for identifying mutations or combinations of mutations that impact epidemic transmission and phenotypic impact can aid the formation of hypotheses that can then be tested using reverse genetics,” Borucki said.
Other Livermore researchers include Mona Hwang, James Thissen, Jonathan Allen and Adam Zemla. The research was funded by LLNL’s Laboratory Directed Research and Development program.
Contact
 Anne M. Stark
Anne M. Stark
[email protected]
(925) 422-9799
Related Links
PLOS ONETags
Physical and Life SciencesFeatured Articles