Research uncovers missing physics in explosive hotspots
 (Download Image)
(Download Image)
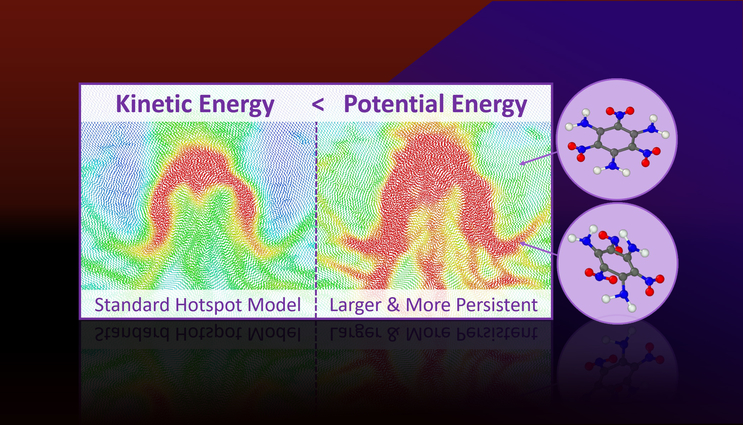
Molecular dynamics simulations predict that more potential energy is localized in hotspots than their kinetic energy (or temperature) would suggest. Excess potential energy is tied to persistent strained molecular states that are primed for chemical reactions and explain why hotspots react faster than the bulk.
Research conducted on Lawrence Livermore National Laboratory’s (LLNL) supercomputer Quartz highlights findings made by scientists that reveal a missing aspect of the physics of hotspots in TATB (1,3,5-trimamino-2,4,6-trinitrobenzene) and other explosives.
Hotspots are localized regions of elevated temperature that form from shock-induced collapse of microstructural porosity and are known to govern the shock initiation and detonation properties of explosives. The main concept behind hotspots is that local elevated temperatures accelerate local chemistry.
The research is featured in the March 11 issue of the Journal of Physical Chemistry Letters and was a collaboration between LLNL and Purdue University. Authors include Matthew Kroonblawd from LLNL and Brenden Hamilton, Chunyu Li and Alejandro Strachan from Purdue.
The work highlights a neglected physical aspect of the early stages of hotspot formation and evolution that provides a route to systematically improve multiphysics models of shock initiation and detonation used to assess performance and safety.
“One of the most puzzling results from early reactive molecular dynamics simulations is that hotspots formed at collapsed pores react much more quickly than ones of equivalent size, temperature and pressure in the bulk material,” Strachan said. “While recognized, the reason behind these differences was not understood. Our study resolves this question in that we find that the explosive material in a collapsed pore is fundamentally different from the bulk and that it is in a high-energy state primed for chemical reactions.”
Importance of understanding hotspots
TATB is an insensitive high explosive critical to the nation’s nuclear stockpile and is challenging to model at the continuum scale. Engineering models for explosive safety and detonation performance rely on physics models that center on the formation and growth of hotspots.
Kroonblawd explained that “continuum-level multiphysics models used to assess safety and performance are highly empirical, which makes it difficult to create explosive models that are transferable to different application conditions. The lack of transferable models is especially true for insensitive high explosives such as TATB. It is still not possible to build an explosive model from first principles, indicating that key aspects are missing from our understanding of hotspot physics and chemistry.”
These models rely on accurate treatments of chemical reactivity and thermal transport; whether hotspots will grow and coalesce into a detonation wave is determined by a competition between the rate of heat generation due to chemistry and heat loss due to thermal conduction.
Identifying the cause behind differences in hotspot reaction rates gives a path toward formulating more general explosive models that will improve their predictive accuracy and transferability. While these models have typically focused on temperature as the main variable controlling chemistry, the findings suggest that recasting these models in terms of the potential energy will yield a more general treatment that can distinguish the differing reactivities of different material states.
Through all-atom molecular dynamics simulations, the researchers found that hotspots are not just regions of localized kinetic energy (or temperature), but are also regions of localized potential energy. The amount of potential energy is much greater than the amount kinetic energy and it is concentrated into molecular modes relevant to chemical decomposition.
The potential energy localization manifests due to molecular-level strains in plastically deformed regions of the material and this will lead to a mechanochemical acceleration of reactions.
“The key takeaway is that there is no one-to-one relationship between kinetic and potential energy in these systems, hence, one cannot infer local reaction rates from only the temperature field,” Hamilton said.
Team conducts large-scale simulations
The work, conducted by Materials Science Division staff in the LLNL Energetic Materials Center (EMC) and the Materials Engineering Department at Purdue, was supported by LLNL’s Laboratory Directed Research and Development Strategic Initiative Program with Lara Leininger, EMC director, as principal investigator. The work involved running large-scale all-atom simulations on the Livermore Computing machine Quartz, and these simulations were performed using computing time granted through LLNL’s Computational Grand Challenge.
To study the longtime relaxation properties of the kinetic and potential energy in hotspots, the team developed a new method called Shock Trapping Internal Boundaries.
“Generally, shock simulations are limited in time to when a shockwave reaches the downstream simulation boundary, which generates state-altering reflection waves,” Hamilton said. “In our method, we can isolate the hotspot, or any region of interest, preventing reflections from interacting with it to allow continuous study of time evolution.”
This allowed the team to quantify the rates for relaxation of kinetic and potential energy to determine that the potential energy of the hotspot persists after thermal conduction dissipates the kinetic energy.
Molecular dynamics simulations predict that more potential energy is localized in hotspots than their kinetic energy (or temperature) would suggest. Excess potential energy is tied to persistent strained molecular states that are primed for chemical reactions and explain why hotspots react faster than the bulk.
Contact
 Michael Padilla
Michael Padilla
[email protected]
(925) 341-8692
Related Links
Journal of Physical Chemistry LettersTags
ASCEnergetic Materials Center
Advanced Materials and Manufacturing
Materials Science
HPC, Simulation, and Data Science
Computing
Physical and Life Sciences
Featured Articles