Lab scientists help develop new tool for faster, more accurate analysis of chemical bonding
 (Download Image)
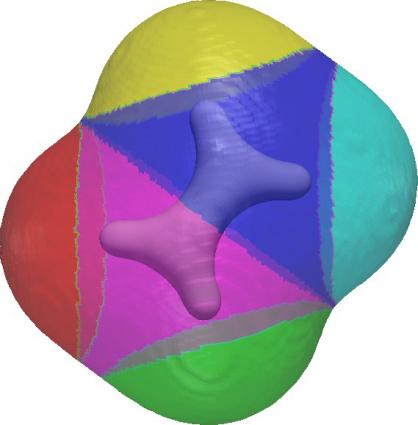
TopoMS, a computational tool enabling detailed topological analysis of molecular and condensed‐matter systems, is highlighted in the June issue of the Journal of Computational Chemistry.
(Download Image)
TopoMS, a computational tool enabling detailed topological analysis of molecular and condensed‐matter systems, is highlighted in the June issue of the Journal of Computational Chemistry.
In a development with the potential to enable breakthroughs in lithium-ion batteries and further impact a wide swath of research areas, Lawrence Livermore National Laboratory (LLNL) scientists have developed a new open source software application that can, in real-time, compute, analyze and potentially predict the trajectories of atoms during the course of bond breaking and formation in dynamical simulations.
The tool, called TopoMS, sprang from a multi-year initiative led by LLNL physicist John Pask, involving researchers from LLNL, Lawrence Berkeley National Laboratory and the University of Utah, and funded by the Department of Energy to devise a new methodology for performing complex quantum molecular dynamics (QMD) simulations on supercomputers. The focus of the recently completed collaboration was to quickly and accurately model solid-liquid interfaces in lithium-ion batteries and ultimately improve energy storage and power.
The project required an approach to topological analysis of molecular and condensed‐matter systems that hadn’t been attempted before, and the result was TopoMS, a new method and code detailed in the June issue of the Journal of Computational Chemistry and featured on the cover.
By combining modern computational science with traditional chemistry and the quantum mechanical theory of atoms in molecules (QTAIM), the researchers said the software could not only lead to a better understanding of lithium-ion battery performance and safety but also could run the gamut of other application areas as well. The research leading to TopoMS includes contributions from computer scientists at both the University of Utah and LLNL.
"This opens up a whole new paradigm of analysis that we couldn’t do before," said LLNL computer scientist Harsh Bhatia, the code’s lead developer and lead author of the paper. "The real power this software brings is consistency compared to other methods. What we did was combine highly accurate numerical algorithms with discrete representations that guarantee that the computational analysis is consistent with the theory of smooth functions."
TopoMS, Bhatia explained, goes a step beyond previous computational tools used to analyze chemical bonding, by leveraging cutting-edge methods and codes from computational topology to robustly and efficiently compute the Morse-Smale Complex (MSC), a mathematical construct used to analyze topology and characterize scalar fields. Previously, the researchers said, many approaches have utilized a crude distance-based analysis that required assumptions about which atoms are bonded and which are not. Because TopoMS, capable of analyzing tens of thousands of atoms at once, makes no such assumptions and is founded upon by the QTAIM, it produces a much more robust and transferable analysis, the researchers said.
"We haven’t invented the idea to understand bonding from the topology of the electron density. What we did was realize that the well-developed theory and machinery of the MSC had the potential to work extremely well to understand our particular scalar field; and in so doing, understand the chemistry and dynamics in lithium-ion batteries," said Pask, director of the DOE SciDAC effort under which TopoMS was developed. "It’s faster, it’s more robust and more accurate than anything that’s come before. It’s elegant mathematically and well suited to parallel implementation. To robustly handle the very large, discrete datasets produced by quantum simulations is a really big deal. It could well become the default tool for this kind of work in the full range of applications areas."
TopoMS is so fast, the researchers said, that it might even be run while simulations are being performed, so scientists can watch in real-time which chemical bonds form and break. With the ability to study on-the-fly bonding, scientists can now look at understanding why the bonds change, answer questions they haven’t thought of and possibly foresee changes to accelerate simulations.
"Rather than have to simulate one system for a nanosecond, which takes very many steps, if you have some way of predicting the likelihood of a certain trajectory, you might just say 'wait, I’ll go this way,'" Pask explained. "Now you can branch off into possible futures, in parallel, and get relevant statistics without having to run the equivalent single, very long simulation."
Although originally aimed at understanding bonding in lithium-ion systems, the researchers said that TopoMS could have broad scientific implications. It has been successfully demonstrated, made publicly available, and is already gathering interest outside the Lab. The next step, the researchers said, is to integrate TopoMS with widely used simulation codes, such as the Vienna Ab initio Simulation Package (VASP), a standard code used for atomic-scale materials modelling, so scientists everywhere can begin to benefit from the new capability.
"A large fraction of computational resources worldwide are used by VASP alone. It would have a huge impact to interface with such a code," Pask said. "It could accelerate so much discovery."
Other key contributors to the TopoMS code included LLNL computer scientist Peer-Timo Bremer, Vincenzo Lordi and the University of Utah’s Attila Gyulassy and Valerio Pascucci. The TopoMS tool is under dual license with the University of Utah and available for download on GitHub.
Contact
 Jeremy Thomas
Jeremy Thomas
[email protected]
(925) 422-5539
Related Links
Discontinuous methods for accurate, massively parallel quantum molecular dynamics: Lithium Ion interface dynamics..."Journal of Computational Chemistry
TopoMS tool
Tags
EngineeringPhysical and Life Sciences
Featured Articles